...
Use the excel to open the files Merged.txt, haplocheck_results (contains the contamination status), andhaplogroups_workshopsamples.txt (contains the haplogroup information for each one of the samples).
Filtering out
...
variants from samples identified as contaminated
- By using the haplocheck_results file, you will check which samples are contaminated (column B: Contamination Status). If there is any sample indicating YES in the contamination status column, you will need to copy the Sample IDs (column A: Sample) and paste in a new excel file. Name Name your file as samples_to_remove and save it as txt format (see the image below as an example).
...
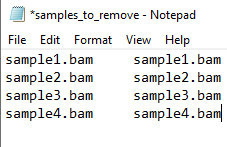
#NOTE: The samples_to_remove.txt file has should have two columns containing and no header. Both columns have the same Sample ID in both columns and no headerinformation (Sample IDs). This file is going to be used in further steps. If you want, you can already upload this file into your work directory in the SCC cluster.
- Continue the next steps using the txt copy file in excel. Check out at the end of this section an example of how the excel file was organized in different sheets based on the QC steps (Figure 1).
Homoplasmic and common variants filtering using PLINK in the SCC cluster
...
module load bio/PLINK/1.9b_6.22-x86_64 |
|---|
#NOTE: The samples_to_remove.txt file was created at the QC section.
Homoplasmic variants calling
...